
Despite the availability of direct-acting antiviral oral combination treatment regimens and eradication efforts by the World Health Organization and others, HCV continues to be a serious viral disease. An estimated 50 million people globally live with chronic HCV infection, with approximately 1 million new infections occurring each year. In 2022, HCV led to an estimated 242,000 deaths. These deaths are primarily attributable to cirrhosis and hepatocellular cancer, each of which are serious long-term consequences resulting from prolonged exposure, generally up to 20 years of HCV infection.
In the US, it is estimated that there up to 4 million persons living with untreated with HCV infection. Further it is reported that on an annual basis in the US, there are approximately 160,000 newly reported chronic HCV infections. This incidence of newly reported infections substantially outpaces rates of treatment, which is estimated to be approximately 85,000 annually.
In the US and elsewhere, HCV is increasingly affecting younger people, with high case rates among those between 20 to 49 years of age. Since recently infected and younger patient populations are less likely to have developed cirrhosis given the relatively shorter cumulative exposure to the virus, there has been a trend in the US of a decreasing incidence of cirrhosis among individuals with HCV infection. It is estimated that less than 10% of the HCV-infected population in the US has cirrhosis.
In 2025, global net sales of HCV therapeutics known in the US as Mavyret® and Epclusa®, together with the authorized generic copy of Epclusa, is approximately $2.5 billion with the US contributing approximately 50% of these sales. Given the large number of patients currently infected with HCV and high rates of annual incidence, it is expected that a substantial global market will exist for the foreseeable future.
The regimen of bemnifosbuvir and ruzasvir has demonstrated a potential best-in-class profile that combines the most compelling attributes of current HCV drug treatments. Atea’s combination regimen is protease inhibitor-free with a profile of short treatment duration, low risk of drug-drug interactions and convenience with no food effect.
The global Phase 2 study enrolled 275 treatment-naïve patients, both with and without compensated cirrhosis. The study was designed to evaluate the safety and efficacy of eight weeks of treatment with the regimen consisting of once-daily bemnifosbuvir 550 mg and ruzasvir 180 mg.
The primary endpoints of the study were safety and sustained virologic response at 12 weeks post-treatment (SVR12) in the per-protocol treatment adherent population. Secondary and other endpoints included SVR12 in the per-protocol population regardless of treatment adherence (efficacy evaluable), virologic failure and resistance.

The global Phase 2 study that evaluating the regimen of bemnifosbuvir and ruzasvir for treatment of HCV met its primary endpoints of safety and SVR12. Primary endpoint results demonstrated a 98% (208/213) SVR12 rate in the per-protocol treatment adherent patient population after eight weeks of treatment with the regimen of bemnifosbuvir and ruzasvir. The efficacy evaluable patient population, which included 17% treatment non-adherent patients, achieved a 95% (242/256) SVR12 rate demonstrating the robust potency and forgiveness of the regimen.


The regimen of bemnifosbuvir and ruzasvir was generally safe and well-tolerated with no drug-related serious adverse events or treatment discontinuations. Results from the Phase 2 study were presented at the European Association for the Study of the Liver (EASL) Congress 2025. You can access the poster presentation here.

In addition to the reported clinical results for the regimen, Atea has also presented preclinical data further demonstrating a high barrier to resistance and favorable pharmacokinetics for bemnifosbuvir and a low risk of drug-drug interactions for ruzasvir. Atea has previously reported a low risk of drug-drug interactions for bemnifosbuvir.
Bemnifosbuvir has been shown in in vitro studies to be approximately 10-fold more active than sofosbuvir (SOF) against a panel of laboratory strains and clinical isolates of HCV GT 1–5. In vitro studies have also demonstrated that bemnifosbuvir remained fully active against SOF resistance-associated substitutions (S282T), with up to 58-fold more potency than SOF. The pharmacokinetic (PK) profile of bemnifosbuvir supports once-daily dosing for the treatment of HCV. Bemnifosbuvir has been shown to have a low risk for drug-drug interactions. Bemnifosbuvir has been administered to over 2,300 subjects and has been well-tolerated at doses up to 550 mg for durations up to 12 weeks in healthy subjects and patients.
Ruzasvir has demonstrated highly potent and pan-genotypic antiviral activity in preclinical (picomolar range) and clinical studies. Ruzasvir has been administered to over 2,100 HCV-infected patients at daily doses of up to 180 mg for 12 weeks and has demonstrated a favorable safety profile. The PK profile of ruzasvir supports once-daily dosing.
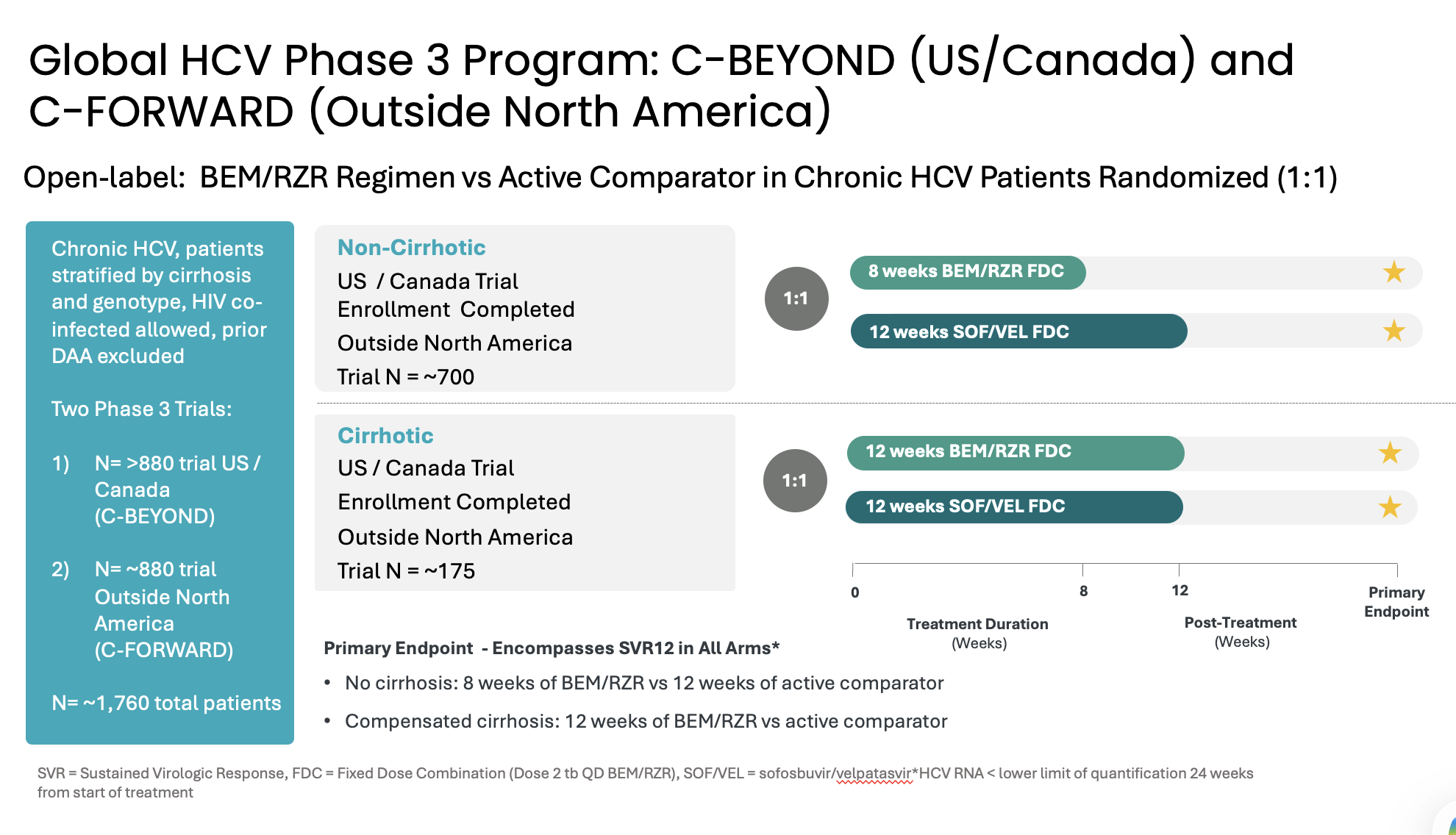
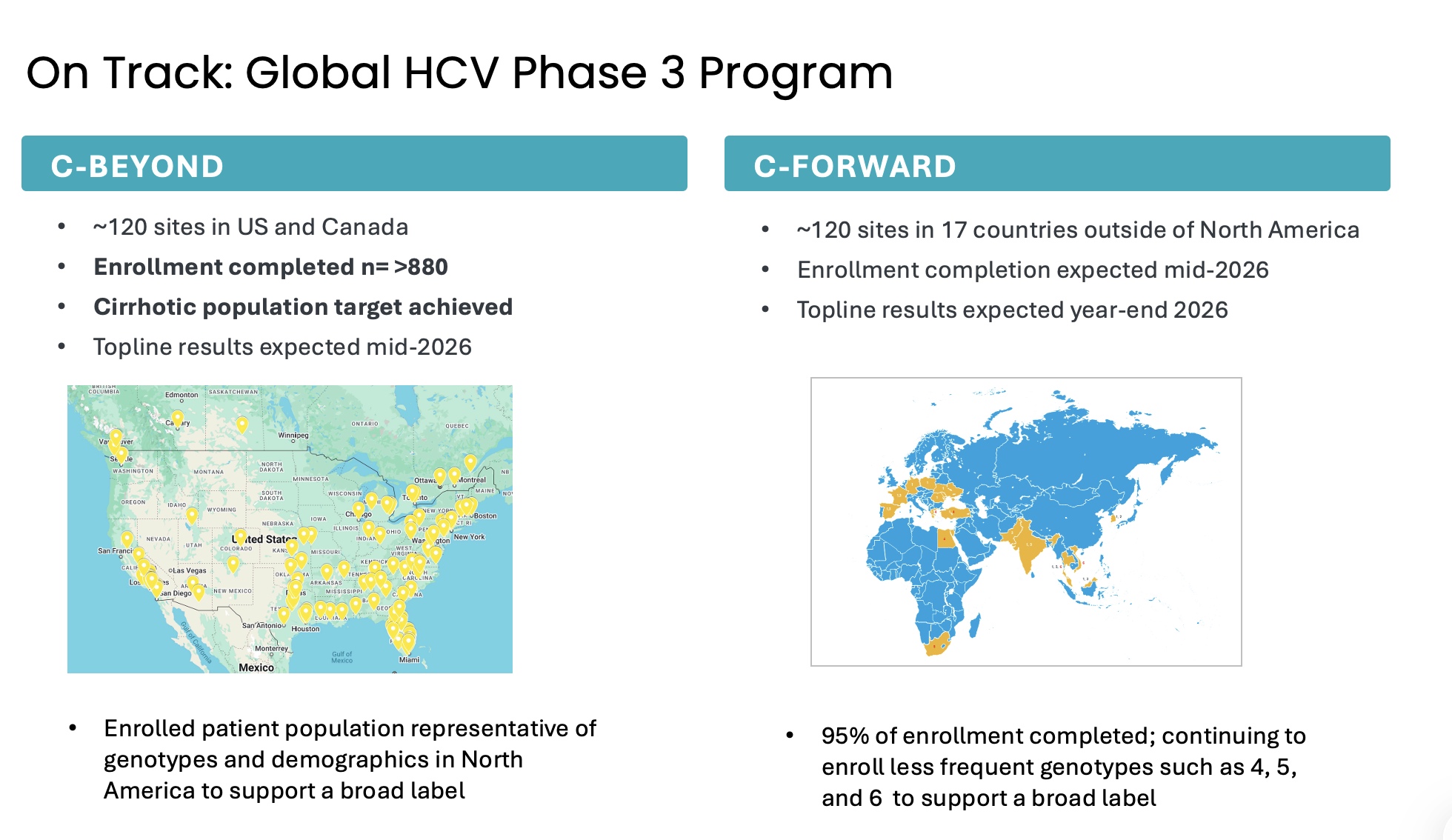
Atea’s HCV Phase 3 development program includes two open-label Phase 3 trials, C-BEYOND being conducted in the US and Canada, and C-FORWARD being conducted outside of North America. Each Phase 3 trial is enrolling approximately 880 treatment-naïve patients, including those with or without compensated cirrhosis. The trials compare the fixed-dose combination (FDC) regimen of bemnifosbuvir and ruzasvir to the FDC regimen of sofosbuvir and velpatasvir. The regimen of bemnifosbuvir and ruzasvir is administered orally once-daily for eight weeks (in patients without cirrhosis) or 12 weeks (in patients with compensated cirrhosis) while the regimen of sofosbuvir and velpatasvir is administered orally once-daily for 12 weeks to all patients, with or without compensated cirrhosis.
The primary endpoint for each trial is HCV RNA < lower limit of quantitation (LLOQ) at 24 weeks from the start of treatment and encompasses sustained virologic response 12 weeks post-treatment (SVR12) in each arm. Measurement at 24 weeks from the start of treatment is to ensure the primary endpoint measurement occurs at the same relative timepoint from the start of treatment in all patients. The primary endpoint will be assessed in the modified intent-to-treat population in C-BEYOND and in the per-protocol population in C-FORWARD.
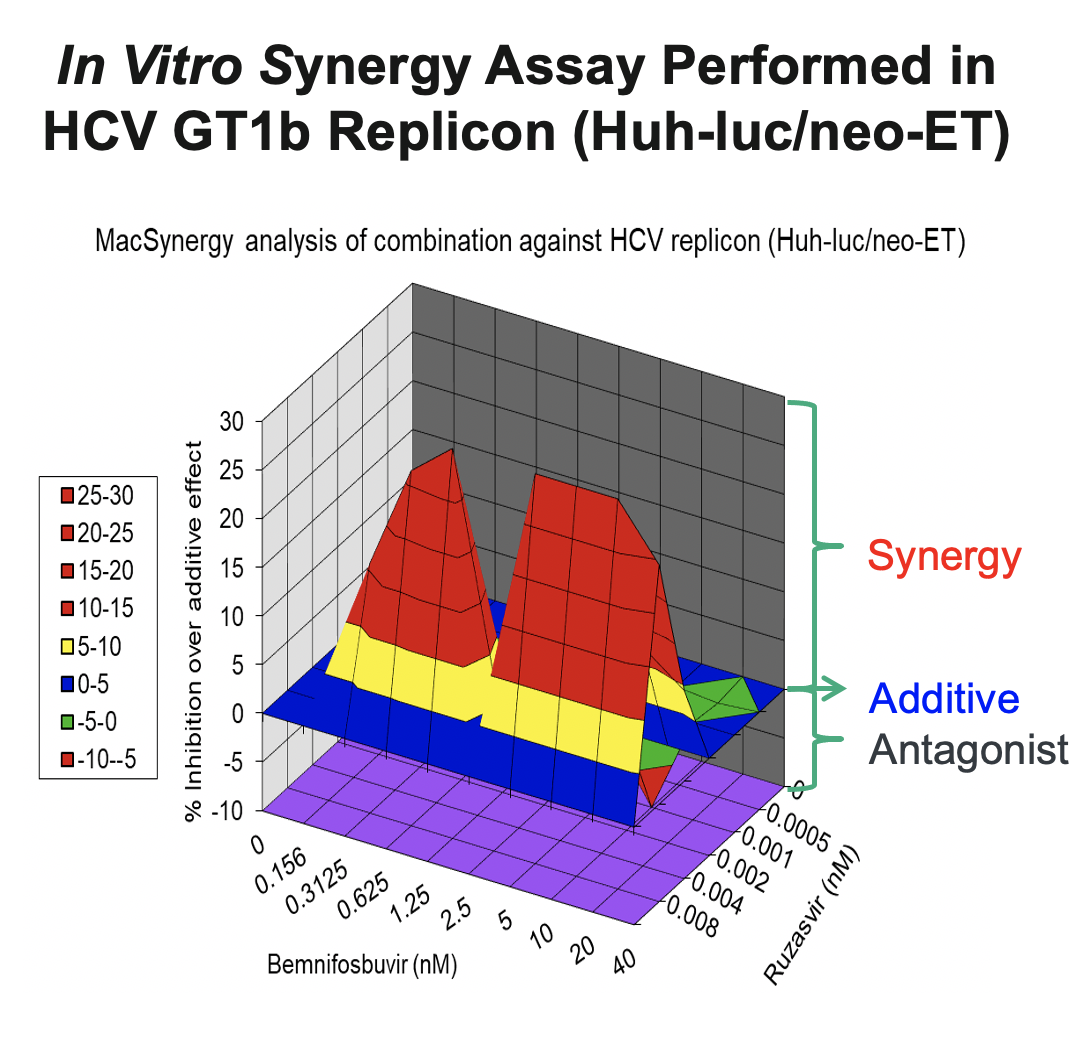
In support of the development of the regimen of bemnifosbuvir and ruzasvir in patients, we conducted in vitro synergy experiments in HCV GT1b replicon assays (Huh-luc/neo-ET), where HCV replicon cells were treated with multiple concentrations of AT-511, the free base of bemnifosbuvir, and ruzasvir either alone or in combination. As shown in the figure below, these experiments demonstrated that the combination resulted in substantially greater inhibition of HCV replication than either agent alone, suggesting a synergistic antiviral effect between the two inhibitors.
Atea is a clinical-stage biopharmaceutical company committed to addressing unmet medical needs through novel antiviral treatments.
Copyright © 2025 By Atea Pharmaceuticals. All Rights Reserved. Privacy Policy | Terms of Use